
Objective: To determine if 80 or 100 mg of Resmetirom compared with placebo resolves NASH and/or reduces fibrosis on liver biopsy and prevents progression to cirrhosis and/or advanced liver disease. Inclusion Criteria: Exclusion Criteria: Participants: 966 Patients Treatment Groups: Primary … Read More
Blog

LIBRETTO-531 Trial: Selpercatinib in Medullay Thyroid Cancer
Objective: To determine if the study drug selpercatinib is safer and more effective compared to standard treatment in participants with RET-mutant medullary thyroid cancer (MTC) that cannot be removed by surgery or has spread to other parts of the body. … Read More

LIBRETTO-431 Trial: Selpercatinib in NSCLC
Objective: To determine if the study drug selpercatinib, compared to a standard treatment, is effective and safe in participants with RET fusion-positive non-squamous non-small cell lung cancer (NSCLC) that has spread to other parts of the body. Inclusion Criteria: Participants: … Read More
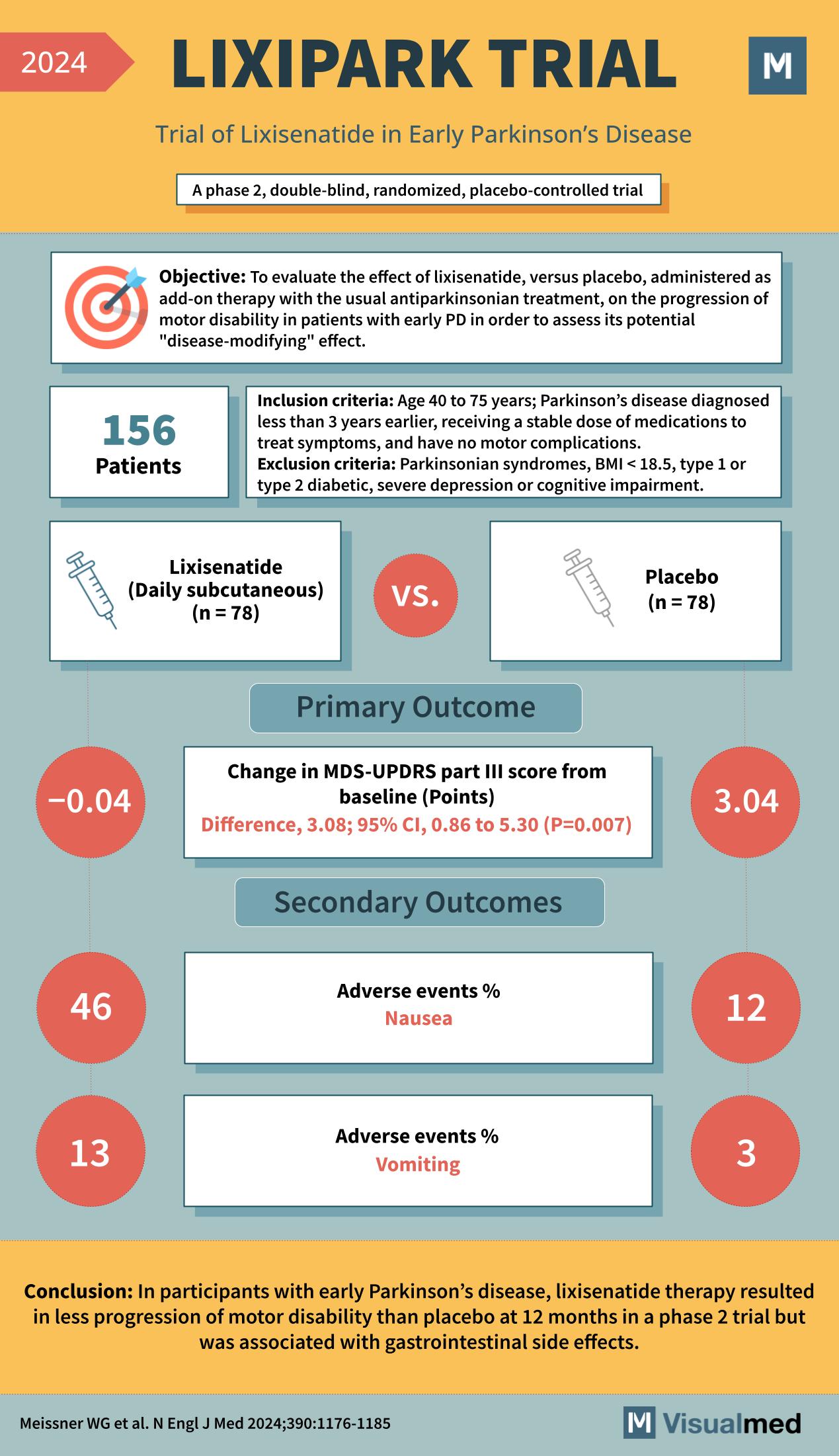
LIXIPARK Trial: Lixisenatide in Early Parkinson’s Disease
Objective: To evaluate the effect of lixisenatide, versus placebo, administered as add-on therapy with the usual antiparkinsonian treatment, on the progression of motor disability in patients with early Parkinson’s Disease (PD) to assess its potential “disease-modifying” effect. Inclusion Criteria: Exclusion … Read More

KETAMORPH Trial: Ketamine for Trauma
Year: 2024 Title: KETAMORPH Subtitle: Ketamine Compared With Morphine for Out-of-Hospital Analgesia for Patients With Traumatic Pain Type of Trial: A prospective, multicenter, single-blind, noninferiority trial Objective: To assess the noninferiority of intravenous ketamine compared with intravenous morphine sulfate to … Read More

CURRENT-OASIS 7: Double Dose Clopidogrel or ASA in ACS
Year: 2010 Title: CURRENT-OASIS 7 Subtitle: Double-dose versus standard-dose clopidogrel and high-dose versus low-dose aspirin in individuals undergoing PCI for ACS Type of Trial: Double-blind, Randomized Factorial Trial Objective: To evaluate whether a higher dosage of clopidogrel with aspirin (two … Read More

FAST-TAVI II: Reducing LOS after TAVI
Year: 2024 Title: FAST-TAVI II Subtitle: Reducing length of stay after transfemoral transcatheter aortic valve implantation Type of Trial: A prospective, cluster, randomized, controlled trial Objective: To evaluate the efficacy and safety of an intervention (a training program) aimed to … Read More

CodeBreak 300 Trial: Sotorasib + Panitumumab in CRC
Year: 2023 Title: CodeBreaK 300 Subtitle: Sotorasib plus Panitumumab in Refractory Colorectal Cancer with Mutated KRAS G12C Type of Trial: A phase 3 randomized, open-label, active-controlled trial Objective: To compare progression-free survival in previously treated participants with KRAS G12C mutated … Read More

VIVA Trial: TAVR in Severe AS and Small Annulus
The infographic summarizes the VIVA TRIAL as follows: Year: 2024 Title: VIVA TRIAL Subtitle: Transcatheter or Surgical Aortic Valve Replacement in Patients With Severe Aortic Stenosis and Small Aortic Annulus Type of Trial: A prospective, randomized, parallel, blinded trial Objective: … Read More

CAROLINA Trial: Linagliptin vs. Glimepiride in T2DM
Year: 2019 Title: CAROLINA TRIAL Subtitle: Effect of Linagliptin vs Glimepiride on Major Adverse Cardiovascular Outcomes in Patients With Type 2 Diabetes Type of Trial: An International, Randomised, Parallel Group, Double Blind Trial Objective: To assess cardiovascular outcomes of linagliptin … Read More